
?")
O PAF-TTR (Polineuropatia Amiloidótica Familiar por TTR), também conhecido como hATTR (Amiloidose hereditária por transtirretina) é uma doença rara e progressiva que faz parte de um grupo de doenças denominadas “Amiloidose”. Este dia é comemorado todo dia 10 de junho.
Por que 10 de junho é o dia do hATTR?
A Polineuropatia Amiloidótica Familiar (PAF) foi descrita em Portugal pelo Dr. Corino Andrade em 1952, daí ser conhecida como Doença de Andrade e a data se deve ao fato do profissional ter nascido em Portugal no dia 10 de junho. Foi ele quem publicou o primeiro artigo com informações baseadas em estudos com pacientes durante um período de 12 anos.
Lá entra em detalhes e os sintomas que conhecemos hoje, além de destacar a informação essencial de que as pessoas acometidas pela Doença de Andrade acumulam a substância anormal em vários locais do corpo, produzindo uma neuropatia mista ao longo dos anos. e progressiva, alterando o funcionamento dos nervos.
O que é polineuropatia amiloidótica familiar?
É uma doença genética rara, progressiva e de difícil diagnóstico. que afeta adultos. O nome polineuropatia sugere uma doença que afeta muitos nervos. É chamada de amiloidose porque seu dano está associado à deposição de fibras proteicas denominadas amilóides, que impactam na estrutura e função dos tecidos afetados.
A TTR (Transtirretina) é uma proteína sintetizada e secretada principalmente pelo fígado e tem como função transportar o hormônio tireoidiano (T4) e a vitamina A. Mutações genéticas alteram o formato dessa proteína, que se dobra de forma anormal, formando fibras rígidas e lineares. (fibrilas amilóides insolúveis) que se acumulam em órgãos e tecidos.
imagem.png
Nesse sentido, o médico César Crespi (MP115.409 / MN 124 765), Hepatologista Centro de Referência para Doenças Raras e Difíceis de Diagnóstico (CERyD) do Hospital San Juan de Dios de La Plata detalha que: “O gene que codifica a proteína transtirretina sofre mutação na amiloidose hereditária, produzindo uma proteína alterada que se depositará nos tecidos e nos dará sintomas específicos relacionados ao acúmulo da substância nos diversos tecidos. Ou seja, em muitos casos, a pessoa afetada herda a doença de um dos pais afetados”, detalha.
A probabilidade de herdar a mutação do progenitor portador é de 50% em cada gravidez e a presença de sinais e sintomas pode variar de acordo com região geográfica ou grupo étnico.
Quais são os sintomas e manifestações?
Na FAP-TTR, as manifestações clínicas são multissistêmicas e debilitantes, com significativo impacto psicológico e familiar. Em relação à descoberta da doença em fases iniciais, Crespi refere-se à faixa dos 25-35 anos e mais avançada nos 50 anos, mas é sempre prudente suspeitar em qualquer caso de Polineuropatia.
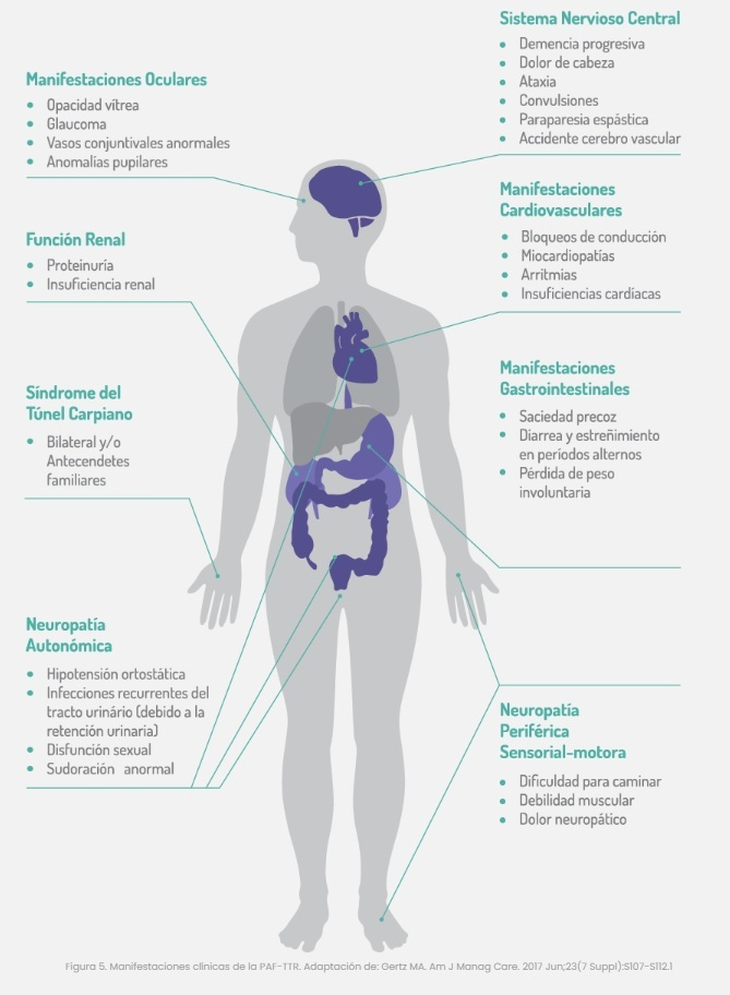
É uma doença rara e de difícil diagnóstico, pois os sintomas, inespecíficos e semelhantes a muitas condições, são frequentemente confundidos com outras entidades clínicas. Podemos encontrar polineuropatia, disfunção gastrointestinal, manifestações oculares, envolvimento da função cardíaca e renal ou síndrome do túnel do carpo (frequentemente bilateral). Pode levar de 3 a 5 anos para chegar a um determinado diagnóstico.
Quanto às características da manifestação, Crespi detalha que “os sintomas iniciais mais incipientes são formigamento e ardência nos membros inferiores (especificamente nos pés) essa sensação vai aumentar – e esclarece – isso se chama manifestação de polineuropatia periférica; A seguir pode-se acrescentar um problema motor, significando que o paciente apresenta dificuldade de mobilidade nos membros inferiores, que evolui para fraqueza muscular, com dificuldade para caminhar até a fase avançada onde o paciente permanece em cadeira de rodas ou até mesmo prostrado.
Neste sentido, o especialista sublinha que “a neuropatia periférica é um desafio diagnóstico porque tem múltiplas causas e manifesta-se também ao nível autonómico, ou seja, naquelas funções independentes da nossa vontade, por exemplo: nos processos digestivos, no controlo do sangue pressão e frequência cardíaca, sudorese, regulação de temperatura. Também pode se manifestar com episódios de taquicardia ou queda da pressão arterial do paciente quando ele para rapidamente”, explica Crespi.
Para que serve um diagnóstico?
Para a abordagem correspondente à ATTRh, o diagnóstico é importante. “Se houver suspeita, a confirmação patológica da deposição de amiloide e o diagnóstico genético são altamente recomendados. Além do mais, Investigações adicionais como avaliação neurológica, cardíaca, autonômica e oftalmológica podem proporcionar mais confiança na obtenção do diagnóstico correto”, afirma o médico. do Hospital San Juan de Dios de La Plata.
“A primeira coisa que se faz quando um paciente chega é um bom exame físico e se tiver sintomas nos membros inferiores, o eletromiograma é um dos estudos mais importantes, o outro estudo relevante é o ecocardiograma (um ultrassom do coração) , e Também pode ser solicitado teste de inclinação (colocar o paciente na maca e mudar de posição e ver como o sistema cardiovascular reage) se as manifestações clínicas o justificarem. Basicamente seria isso e depois dependendo dos sintomas pode-se investigar outros tecidos e órgãos”, explica.
“O teste genético é um componente crucial para determinar a doença. Em pacientes com histórico familiar ou com conjunto de sintomas que representem sinais de alerta. Em famílias com uma mutação conhecida, podem ser realizados testes genéticos diretos para essa mutação. Quando a mutação é desconhecida ou não há histórico familiar, deve-se realizar o sequenciamento completo do gene TTR para identificar a variante genética correspondente”, ressalta.
CComo é obtido um diagnóstico?
Biópsia de tecido. Recomenda-se a confirmação do depósito amiloide através de biópsia tecidual, mas um resultado negativo “não descarta” o diagnóstico, portanto, se a suspeita persistir, devemos continuar estudando os pacientes. Quando a biópsia tecidual é realizada, a presença de depósitos de material amilóide é confirmada pela técnica de coloração Vermelho Congo e avaliação com microscópio de luz polarizada.
O diagnóstico pode ser feito em locais menos invasivos, como biópsia de glândulas salivares menores (mucosa oral), ou extração de gordura subcutânea abdominal por punch (elemento de amostragem mini-invasivo). Na Argentina, estes são os locais mais frequentes para amostragem de tecidos. A sensibilidade da biópsia das glândulas salivares labiais em pacientes com mutação V30M é alta e varia entre 75-91%.
Crespi detalha a importância de acelerar os resultados genéticos nestes tempos e valoriza que “com diagnóstico precoce e medicação adequada, a progressão da doença pode ser retardada. Hoje não tem cura, mas existem opções terapêuticas que permitem deter a progressão da doença”, encerra o profissional.
Texto elaborado com a contribuição de César Crespi (MP115.409 / MN 124 765), Hepatologista Centro de Referência para Doenças Raras e Difíceis de Diagnóstico (CERyD) do Hospital San Juan de Dios de La Plata.

“Fanático hardcore de mídia social. Propenso a ataques de apatia. Criador. Pensador. Guru dedicado da web. Aficionado por cultura pop. Solucionador de problemas.”





